Seurat (Butler et. al 2018) and Scanpy (Wolf et. al 2018) are two great analytics tools for single-cell RNA-seq data due to their straightforward and simple workflow. However, for those who want to interact with their data, and flexibly select a cell population outside a cluster for analysis, it is still a considerable challenge using such tools.
Here we provide a tutorial to help you load the analysis results from Seurat and Scanpy single-cell objects (.rds and .h5ad files) into BioTuring Browser (BBrowser) – to take advantage of the software’s interactive interface and perform downstream analyses on any populations. These include finding marker genes, running sub-clustering and differential expression analysis, comparing cell compositions in different conditions and many other options, all in an interactive and flexible approach.
To support academic research, importing Seurat and Scanpy objects to BBrowser is now free for academic users.
1. What file type does BBrowser support?
BBrowser supports importing Seurat objects (.rds) and Scanpy objects (.h5ad/ h5). For other single-cell object formats, you can convert it to Seurat objects by the tutorial from Satijia Lab.
2. What information does BBrowser collect from the objects?
Upon receiving a Seurat or Scanpy object, BBrowser will read all the data available. If some data are not available in your Seurat/ Scanpy object, BBrowser will run the processing steps based on the latest processed data it can retrieve.
For example, you have a Seurat object with PCA and t-SNE calculated, but not UMAP. When you import the object to BBrowser, if you want to see UMAP, BBrowser will take the PCA to construct UMAP at your request.
Intuitively, well processed and annotated Seurat and Scanpy objects will be submitted in a short time. BBrowser also retains all the metadata of the samples, only if they are characters.
3. How to submit a Seurat or Scanpy single-cell object to BBrowser?
If you have a Seurat/ Scanpy object, simply open BBrowser, go to Data > Add a new study > Other > Drop a file in and click Start to submit the object
Steps to import a Seurat or Scanpy object into BBrowser
We use the “HumanLiver” data set, which is publicly available in the library(HumanLiver) from Bader Lab to demonstrate. You can run the R script below to get the object:
install.packages("devtools")
devtools::install_github("BaderLab/HumanLiver")
library(Seurat)
library(HumanLiver)
viewHumanLiver()
#close the pop-up window
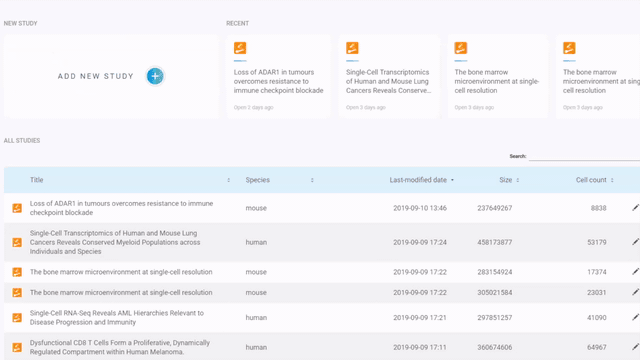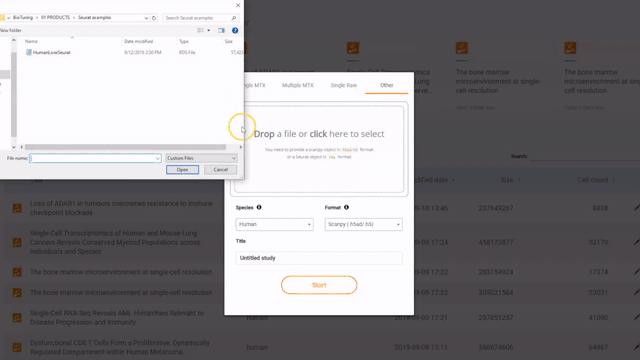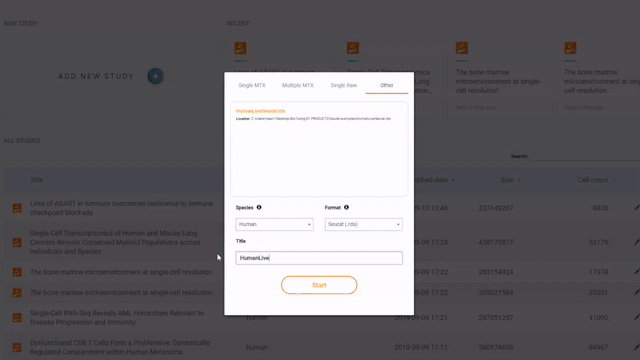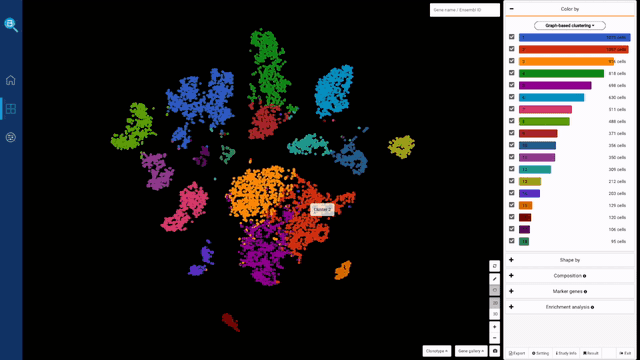
saveRDS(HumanLiverSeurat, file = "C:/Users/BioTuring/Desktop/HumanLiverSeurat.rds")Once submitted to BBrowser, you can click on any cluster and go to Marker genes tab on the right > Find marker genes. Enrichment analysis will be done automatically after marker genes are found.
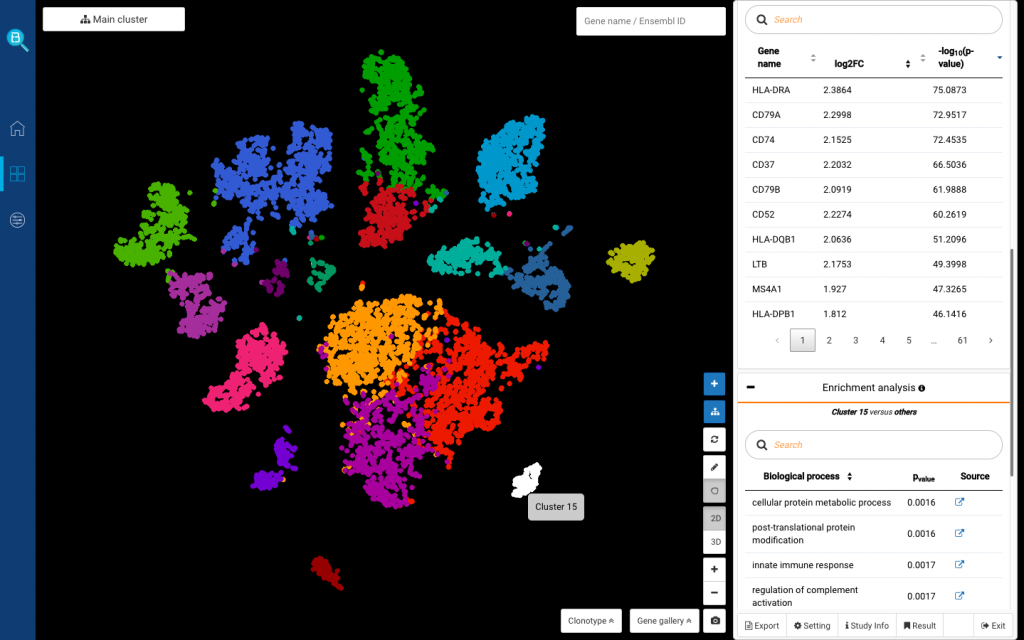
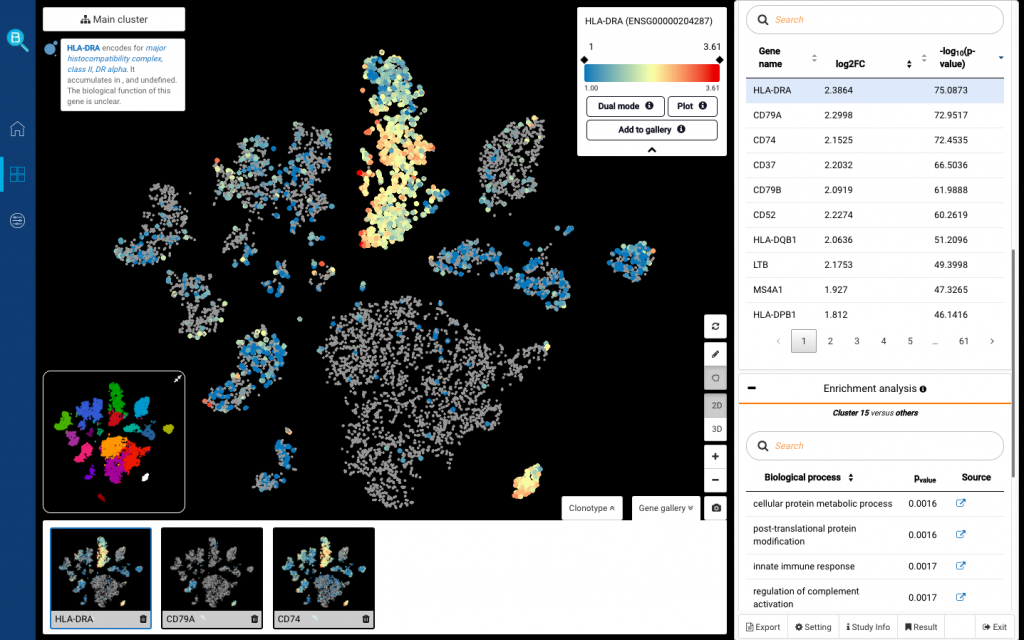
If you click the gene name, BBrowser will show its expression level in the whole population and details of the gene and protein functions. Drop down from the gene query and click Add to Gallery to save the expression plot.
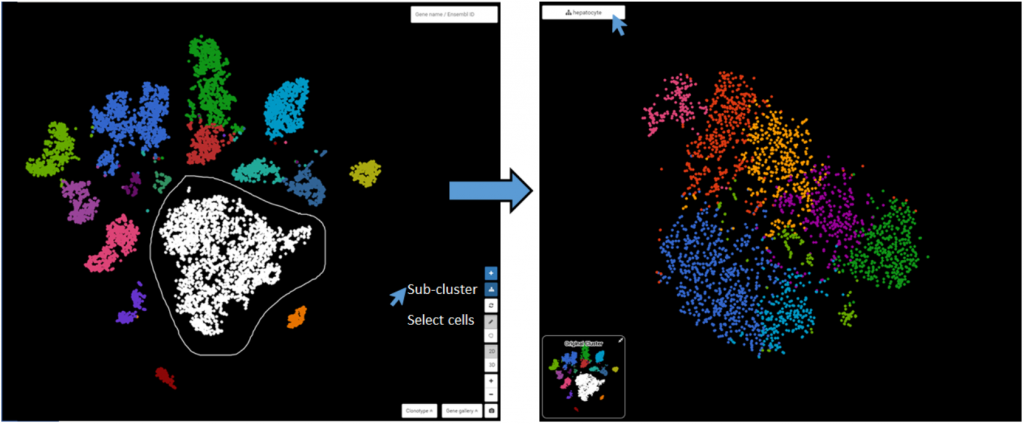
You can also circle any group of cells for marker gene finding and further analysis. Here, we try circling a population and run sub-clustering by clicking on Sub-cluster button. A new sub-clustering dashboard will open, showing the sub-clusters of the population we circled, together with sub-clusters’ marker genes. Click on the hierarchical icon on the top left to go back.
4. Does it accept other single-cell object formats (SingleCellExperiment, Loom,…)?
In case you have another single-cell object format, you can convert it to Seurat and submit to BBrowser.
We use the “manno_human” data set from Martin Hemberg’s group to demonstrate conversion of SingleCellExperiment objects to Seurat objects. You can get the object and the conversion R script here:
# download the dataset using this link
# https://scrnaseq-public-datasets.s3.amazonaws.com/scater-objects/manno_human.rds
library(Seurat)
manno <- readRDS(file = "C:/Users/BioTuring/Desktop/manno_human.rds")
manno.seurat <- as.Seurat(manno, counts = "counts", data = "logcounts")
manno.seurat@assays$RNA@data <- as(manno.seurat@assays$RNA@data, "sparseMatrix")
saveRDS(manno.seurat, file = "C:/Users/BioTuring/Desktop/mannoSeurat.rds")After getting the converted Seurat object, you can submit it to BBrowser by going to Data > Add a new study > Other > Drop a file in and click Start.
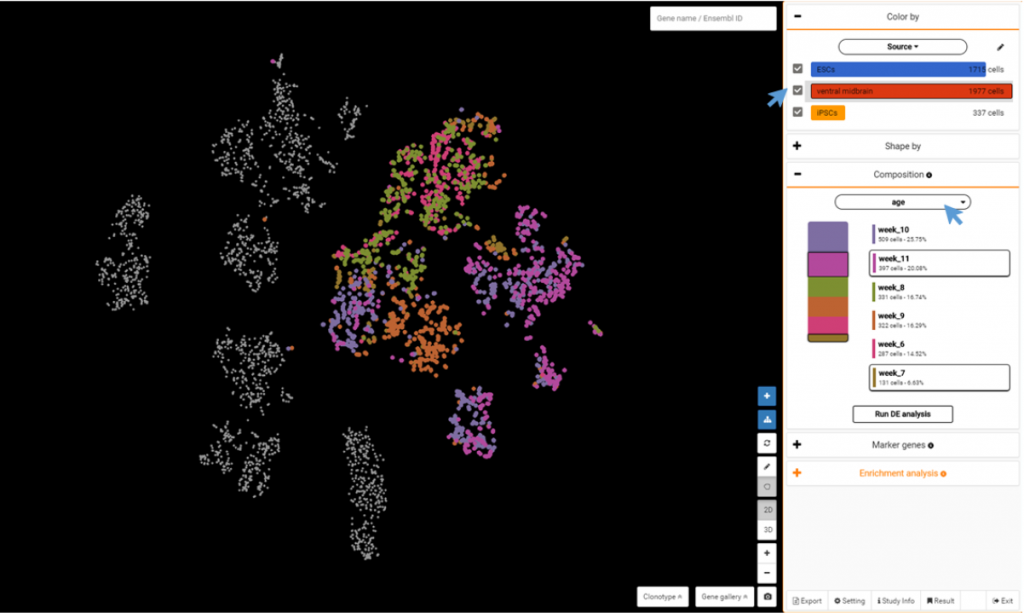
For this dataset, you can color the population by source (ESCs, ventral midbrain, iPSCs) by clicking the drop down from Color by > Your annotation > Source.
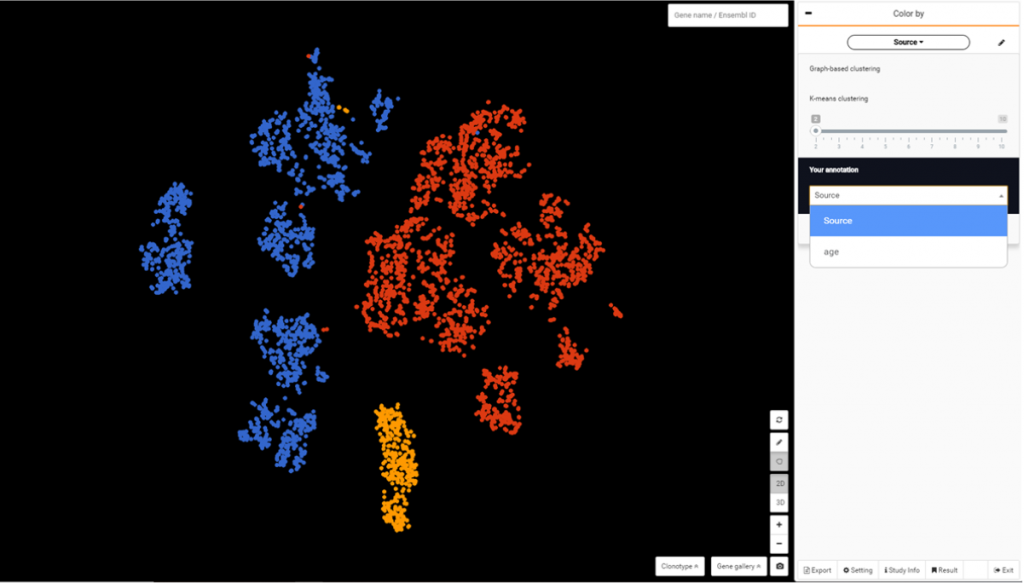
Choose a cluster and go to Composition to view percentage of cells by age. Select any 2 groups by age and click to Run DE analysis.
Above are a few simple steps to load Seurat and Scanpy objects into BBrowser for interactive visualization and analysis. You can also check out the video tutorial below if it is easier to follow:
Again, importing Seurat and Scanpy objects is now free for academic users. If you are interested, download BBrowser at: https://bioturing.com/bbrowser
Feel free to leave your questions in the comment box below, or contact us at support@bioturing.com. We would be happy to help.
Natalia Torow
I have difficulties importing a Seurat object (.rds file) and get the message that the file format could not be recognized. Do you have any suggestions what i could try?
Thanks!
biomembers
Hi Natalia, thank you for letting us know. Our support team will reach out to you soon.
Zerina
Hello,
I am trying to upload my Seurat object (which I exported with saveRDS() ) to Bioturing but I get the following message:
Error in .requirePackage(package) : unable to find required package “SeuratObject”
How can I fix it?
Many thanks!
AS
Getting the same error for an .rds file. Error in .requirePackage(package) : unable to find required package “SeuratObject”
Holger
Getting the same error for an .rds file. Error in .requirePackage(package) : unable to find required package “SeuratObject” . Using in R SeuratObject_4.0.0 and Seurat_4.0.1. Had no problems with Seurat<4.0.
biomembers
Hi Holger, currently BBrowser directly supports Seurat 3 objects. For later versions, you can use our open-source package rBCS (https://github.com/bioturing/rBCS) to convert the objects into a .BCS file and load the BCS file into BBrowser.
Sumgar
Hi! Why does BBrowser not have a linux build?