Assigning cell type identity to cells is a basic yet vital step required in single-cell RNA Sequencing data analysis (scRNA-Seq), often done after dimensionality reduction and scRNA-Seq clustering . If you have successfully captured informative clusters, it’s time to face an even harder challenge: identify what cell type or cell state that a cell population represents.
The sad news is that there’s no one-size-fits-all solution. Cell type annotation is a data-specific task that the scientific community has not agreed on a standard workflow.
Nevertheless, in this article, we’ll summarize common approaches and tools in assigning cell type identity to cells, from which readers can design the optimal workflow for their dataset. We’ll also show you how the comprehensive toolbox of BBrowser can support whichever approach you embrace.
Before we go into details, let’s discuss a vital question:
What Level of Cell “Type” Are We Looking For?
The first and foremost challenge in assigning cell type identity to cells is to define what is the sort of “cell type” (or more accurately, “cell identity”) that we’re looking for.
The definition of “cell type” is not consensus for two reasons. One, a cell can be categorized at many levels from tissue to molecular, or by different criteria. For example, immune cells can be further divided into T cells, B cells, natural killer cells, etc. Within each group, say T cells, surface markers and specific functions distinguish them into more subtypes (CD4+ T cells, CD8+ T cells, T regulatory, etc.). Depending on how deep and specific that you want to divide cell populations, the definition of “cell type” varies.
The second reason is our incomplete understanding about cell identities. As we zoom closer into cellular heterogeneity, novel cell subtypes are being discovered. Often, the transcriptomic profiles of these cells differ not so much that it poses a great challenge for us to draw the border. Cells existing in a gradient of states is also a common phenomenon in developing tissues.
Due to these problems, before annotating cell populations, the researcher must decide what level of “cell identity” that he is looking for. This question is also important for the clustering step, since it gives users a rough estimation of the right number of clusters.
However, splitting the dataset too much from the beginning is not always wise. Too many clusters can make annotation more challenging, especially for very heterogeneous datasets. A useful trick is to use sub-clustering: users can first cluster and annotate at a moderate level, then take out some interesting clusters and treat it as a new dataset. By focusing on just a subset of the data, the resolution will increase and interesting cell subtypes can be explored more easily (Figure 1).

Figure 1. BBrowser Sub-clustering function reveals more clusters within the Monocytes population
Two Common Approaches: Manual or Automated?
There are currently two options in scRNA-Seq cell type annotation: (1) cell populations are examined manually by researchers to define their identities based on marker features, or (2) automated annotation tools analyze the transcriptomics profiles to assign cell identities.
Each option has their pros and cons. While manual curation is time-consuming, demands a strong biological understanding, and is prone to bias, it’s very flexible to adapt with specific datasets, especially those containing rare cell types. On the other hand, automated tools relieve researchers with speed and capacity, but users must be careful with the suitability – not all datasets can be accurately annotated by automatic methods.
2.1. Automated tools for scRNA-Seq cell type annotation
The number of automated tools to solve scRNA-Seq cell identities is increasing. To date, most tools embrace one of three following principles (Figure 2):
- Marker gene databases-based: cells are identified through known marker genes, taken from a reference, such as cell type atlases or published studies. Algorithms will score and rank clusters based on the expression level of marker genes, from which clusters get assigned to a cell type. Some popular tools in this category include SCINA (Zhang et al., 2019) and CellAssign (Zhang et al., 2019).
- Correlation-based: in this approach, clusters are queried against a reference. The algorithm calculates how much each cluster correlates to a cell type in the reference. A cluster is annotated to the best correlated cell type found from the reference. Frequently used tools in this category are Seurat v4 Reference Mapping (Hao et al., 2021), SingleR (Aran et al., 2019), scmap-cluster (Kiselev et al., 2018), etc.
- Supervised classification-based: tools in this category are machine learning algorithms. First, the tool needs to be trained with annotating reference scRNA-Seq datasets. Then, researchers can use it to query their datasets. Garnett (Pliner et al., 2019), scPred (Alquicira-Hernandez et al., 2019), scClassify (Lin et al., 2020) are some noteworthy names in this category.
Readers who are interested in learning about these automated tools in more details can refer to an excellent review from Pasquini et al., (2021)
BioTuring has launched a supervised classification-based tool called Talk2Data Cell Type Prediction. Our algorithm is trained with a huge, curated database containing millions of cells. Our benchmark study shows that BioTuring Cell Type Prediction achieves identical, if not superior performance, in assigning cell type identity to cells when compared to other popular tools (unpublished data).
For instance, we annotated the liver cancer T cell populations from Zheng et al., 2017 using BioTuring Cell Type Prediction and Seurat v4 (Figure 3). Both tools identified major T cell types, such as naive CD4+ T cell, central memory CD4+ T cell (CD4 TCM), effector memory CD4+ T cell (CD4 TEM), regulatory CD4+ T cell (CD4 Treg), naive CD8+ T cell, and mucosal associated invariant CD8+ T cell (CD8 MAIT).
However, while Seurat v4 incorrectly annotated effector memory CD8+ T cell with cytotoxic CD4+ T cell and NK cell, BioTuring algorithm correctly identified them and detected more subtypes, such as: exhausted CD4+ T cell, exhausted CD8+ T cell, NKT-like CD8+ cell.
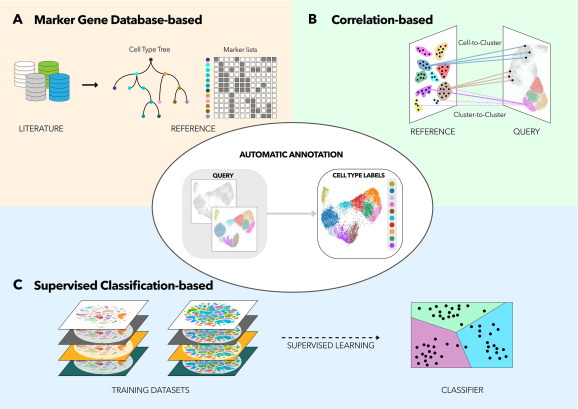
Figure 2. Three types of Automated tools for scRNA-Seq cell type annotation (originally Figure 1, Pasquini et al., 2021)
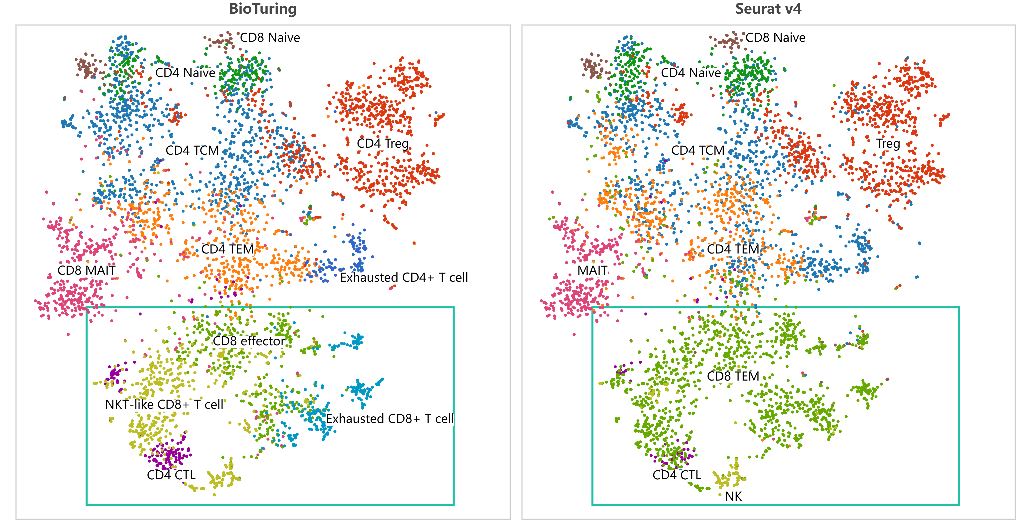
Figure 3. Comparing BioTuring Cell Type Prediction versus Seurat v4 in annotating a dataset from Zheng et al., 2017 for liver cancer T cells. Both tools return highly similar results, but BioTuring identified more cell subtypes
2.2. Manual scRNA-Seq cell type annotation
The principle of manual curation is always identifying cells through marker genes. In BBrowser, this task can be done through the Marker Features function or by Differential Expression (DE) analysis of a cell population against the rest of a dataset.
In the easiest case, when most cells in a cell population highly express canonical marker genes for a cell type, the annotation is almost done. However, in reality and in case of rare cell types, classic marker genes are not enough to identify or distinguish cell populations. Lesser known marker genes need to be examined.
But where do we find marker genes? The ideal reference is a single-cell atlas, preferably if the sample comes from a similar biological context (organism/ tissue/ disease, etc.) Some trustworthy resources are summarized here. Nevertheless, literature mining is recommended, especially if your dataset covers cell types that have not been investigated before.
What if some cells express little to no known marker genes? Unfortunately, this is not an uncommon scenario. There are two possibilities: said cells could be of poor quality or belong to a novel cell type. An expert curator would proceed with enrichment analysis to find what functions cells in these strange clusters perform.
Besides the said functions, BBrowser makes manual annotation more of a breeze with the two following functions: Cell Type Prediction and Cell Search Engine. With the former function, users only need to submit a list of marker genes as the reference. Whenever you select a cell population, BBrowser will automatically query the expression patterns of the reference on the selected cells. The annotation is announced if there is a match between the reference and the selected cells.
The latter, Cell Search Engine, is our out-of-the-box solution where we re-formulate the task: instead of trying to define the cell types, the engine searches for similar cell populations from studies in the BioTuring public database, returning not only the suggested cell type, but also similar signature genes and enriched processes.
How to Know if You’re On the Right Track? Always Validate Your Annotation
None of the methods mentioned above is fool-proof. At its core, the transcriptomic profiles reflect just a part of cell identities so cell type annotation exclusively based on scRNA-Seq analysis may not be accurate.
A handful of validation methods exist. For instance, Chung et al. proposed a non-parametric method to test the statistical significance of cluster membership. By checking the clustering result, the annotation, which is largely influenced by clustering, can be validated (Chung et al., 2020).
In a different approach, combining your scRNA-Seq with other experimental methods is recommended. It is also required if you detect a novel cell type and want to confirm it. Some single-cell techniques that can complement your annotation are Cellular Indexing of Transcriptomes and Epitopes by Sequencing (CITE-seq), spatial transcriptomics, single-cell immune profiling, etc.
Take Away
- Before cell type annotation, decide what level of “cell identity” that you want to find.
- Use sub-clustering to increase resolution in interesting subsets.
- Manual curation is time-consuming, demands a strong biological understanding, and is prone to bias, but it’s very flexible and more reliable to detect rare cell types or with highly heterogenous cell populations.
- Automated tools are fast but are heavily influenced by available references. A good reference / training dataset should be as similar as possible to the queried cell population in terms of biological context.
- When describing novel cell types, experimental validation is a must. Using a multi-omics approach also helps validating cell type annotation as the transcriptomes only reflect a part of a cell’s identity.
Despite no mutual agreement in performing scRNA-Seq cell type annotation, researchers can still enjoy a plethora of supporting tools. It’s probably wise to use a combined approach: start with an automated tool to get a general description of your cell identities, then cross-check with manual curation and validation methods (Clarke et al., 2021).
Reference:
- Zhang, Z., Luo, D., Zhong, X., Choi, J. H., Ma, Y., Wang, S., … & Wang, T. (2019). SCINA: a semi-supervised subtyping algorithm of single cells and bulk samples. Genes, 10(7), 531.
- Zhang, A. W., O’Flanagan, C., Chavez, E. A., Lim, J. L., Ceglia, N., McPherson, A., … & Shah, S. P. (2019). Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nature methods, 16(10), 1007-1015.
- Hao, Y., Hao, S., Andersen-Nissen, E., Mauck III, W. M., Zheng, S., Butler, A., … & Satija, R. (2021). Integrated analysis of multimodal single-cell data. Cell, 184(13), 3573-3587.
- Aran, D., Looney, A. P., Liu, L., Wu, E., Fong, V., Hsu, A., … & Bhattacharya, M. (2019). Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature immunology, 20(2), 163-172.
- Kiselev, V. Y., Yiu, A., & Hemberg, M. (2018). scmap: projection of single-cell RNA-seq data across data sets. Nature methods, 15(5), 359-362.
- Pliner, H. A., Shendure, J., & Trapnell, C. (2019). Supervised classification enables rapid annotation of cell atlases. Nature methods, 16(10), 983-986.
- Alquicira-Hernandez, J., Sathe, A., Ji, H. P., Nguyen, Q., & Powell, J. E. (2019). scPred: accurate supervised method for cell-type classification from single-cell RNA-seq data. Genome biology, 20(1), 1-17.
- Lin, Y., Cao, Y., Kim, H. J., Salim, A., Speed, T. P., Lin, D. M., … & Yang, J. Y. H. (2020). scClassify: sample size estimation and multiscale classification of cells using single and multiple reference. Molecular systems biology, 16(6), e9389.
- Pasquini, G., Arias, J. E. R., Schäfer, P., & Busskamp, V. (2021). Automated methods for cell type annotation on scRNA-seq data. Computational and Structural Biotechnology Journal, 19, 961-969.
- Zheng, C., Zheng, L., Yoo, J. K., Guo, H., Zhang, Y., Guo, X., … & Zhang, Z. (2017). Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell, 169(7), 1342-1356.
- Chung, N. C. (2020). Statistical significance of cluster membership for unsupervised evaluation of cell identities. Bioinformatics, 36(10), 3107-3114.
- Clarke, Z. A., Andrews, T. S., Atif, J., Pouyabahar, D., Innes, B. T., MacParland, S. A., & Bader, G. D. (2021). Tutorial: guidelines for annotating single-cell transcriptomic maps using automated and manual methods. Nature protocols, 16(6), 2749-2764.